Load Data.
data(ehr_data) data <- PhecapData(ehr_data, "healthcare_utilization", "label", 0.4, patient_id = "patient_id") data
## PheCAP Data
## Feature: 10000 observations of 587 variables
## Label: 119 yes, 62 no, 9819 missing
## Size of training samples: 109
## Size of validation samples: 72Specify the surrogate used for surrogate-assisted feature extraction (SAFE). The typical way is to specify a main ICD code, a main NLP CUI, as well as their combination. In some cases one may want to define surrogate through lab test. The default lower_cutoff is 1, and the default upper_cutoff is 10. Feel free to change the cutoffs based on domain knowledge.
surrogates <- list( PhecapSurrogate( variable_names = "main_ICD", lower_cutoff = 1, upper_cutoff = 10), PhecapSurrogate( variable_names = "main_NLP", lower_cutoff = 1, upper_cutoff = 10), PhecapSurrogate( variable_names = c("main_ICD", "main_NLP"), lower_cutoff = 1, upper_cutoff = 10))
Run surrogate-assisted feature extraction (SAFE) and show result.
system.time(feature_selected <- phecap_run_feature_extraction(data, surrogates))
## user system elapsed
## 243.326 0.233 244.236
feature_selected## Feature(s) selected by surrogate-assisted feature extraction (SAFE)
## [1] "main_ICD" "main_NLP" "NLP56" "NLP93" "NLP160" "NLP161" "NLP306"
## [8] "NLP403" "NLP536"Train phenotyping model and show the fitted model, with the AUC on the training set as well as random splits
suppressWarnings(model <- phecap_train_phenotyping_model(data, surrogates, feature_selected)) model
## Phenotyping model:
## $lasso_bic
## (Intercept) main_ICD main_NLP
## 0.8165615 2.3789729 2.7761557
## main_ICD&main_NLP healthcare_utilization NLP56
## -1.6390179 -1.3316767 0.0000000
## NLP93 NLP160 NLP161
## -1.6091941 0.0000000 0.0000000
## NLP306 NLP403 NLP536
## 0.0000000 0.0000000 0.9910775
##
## AUC on training data: 0.988
## Average AUC on random splits: 0.971Validate phenotyping model using validation label, and show the AUC and ROC
validation <- phecap_validate_phenotyping_model(data, model)
## Warning in regularize.values(x, y, ties, missing(ties)): collapsing to unique
## 'x' values
## Warning in regularize.values(x, y, ties, missing(ties)): collapsing to unique
## 'x' values
validation## AUC on validation data: 0.867
## AUC on training data: 0.988
## Average AUC on random splits: 0.971round(validation$valid_roc[validation$valid_roc[, "FPR"] <= 0.2, ], 3)
## cutoff pos.rate FPR TPR PPV NPV F1
## [1,] 1.000 0.007 0.000 0.245 1.000 0.353 0.394
## [2,] 0.988 0.319 0.000 0.356 1.000 0.390 0.526
## [3,] 0.975 0.361 0.000 0.468 1.000 0.436 0.637
## [4,] 0.966 0.375 0.048 0.545 0.965 0.463 0.697
## [5,] 0.959 0.375 0.048 0.603 0.969 0.497 0.743
## [6,] 0.936 0.458 0.048 0.661 0.971 0.536 0.786
## [7,] 0.861 0.542 0.048 0.718 0.973 0.582 0.827
## [8,] 0.785 0.569 0.048 0.776 0.975 0.637 0.864
## [9,] 0.772 0.583 0.095 0.784 0.952 0.633 0.860
## [10,] 0.770 0.583 0.095 0.784 0.952 0.633 0.860
## [11,] 0.767 0.583 0.095 0.784 0.952 0.633 0.860
## [12,] 0.764 0.583 0.095 0.784 0.952 0.633 0.860
## [13,] 0.762 0.597 0.143 0.785 0.930 0.621 0.851
## [14,] 0.756 0.597 0.143 0.789 0.931 0.626 0.854
## [15,] 0.750 0.597 0.143 0.793 0.931 0.630 0.856
## [16,] 0.742 0.611 0.143 0.797 0.931 0.635 0.859
## [17,] 0.732 0.611 0.143 0.801 0.932 0.640 0.861
## [18,] 0.718 0.625 0.190 0.804 0.911 0.630 0.854
## [19,] 0.696 0.625 0.190 0.804 0.911 0.630 0.854
## [20,] 0.674 0.625 0.190 0.804 0.911 0.630 0.854
## [21,] 0.652 0.625 0.190 0.804 0.911 0.630 0.854
## [22,] 0.631 0.625 0.190 0.804 0.911 0.630 0.854phecap_plot_roc_curves(validation)
## Warning: Use of `df$value_x` is discouraged. Use `value_x` instead.## Warning: Use of `df$value_y` is discouraged. Use `value_y` instead.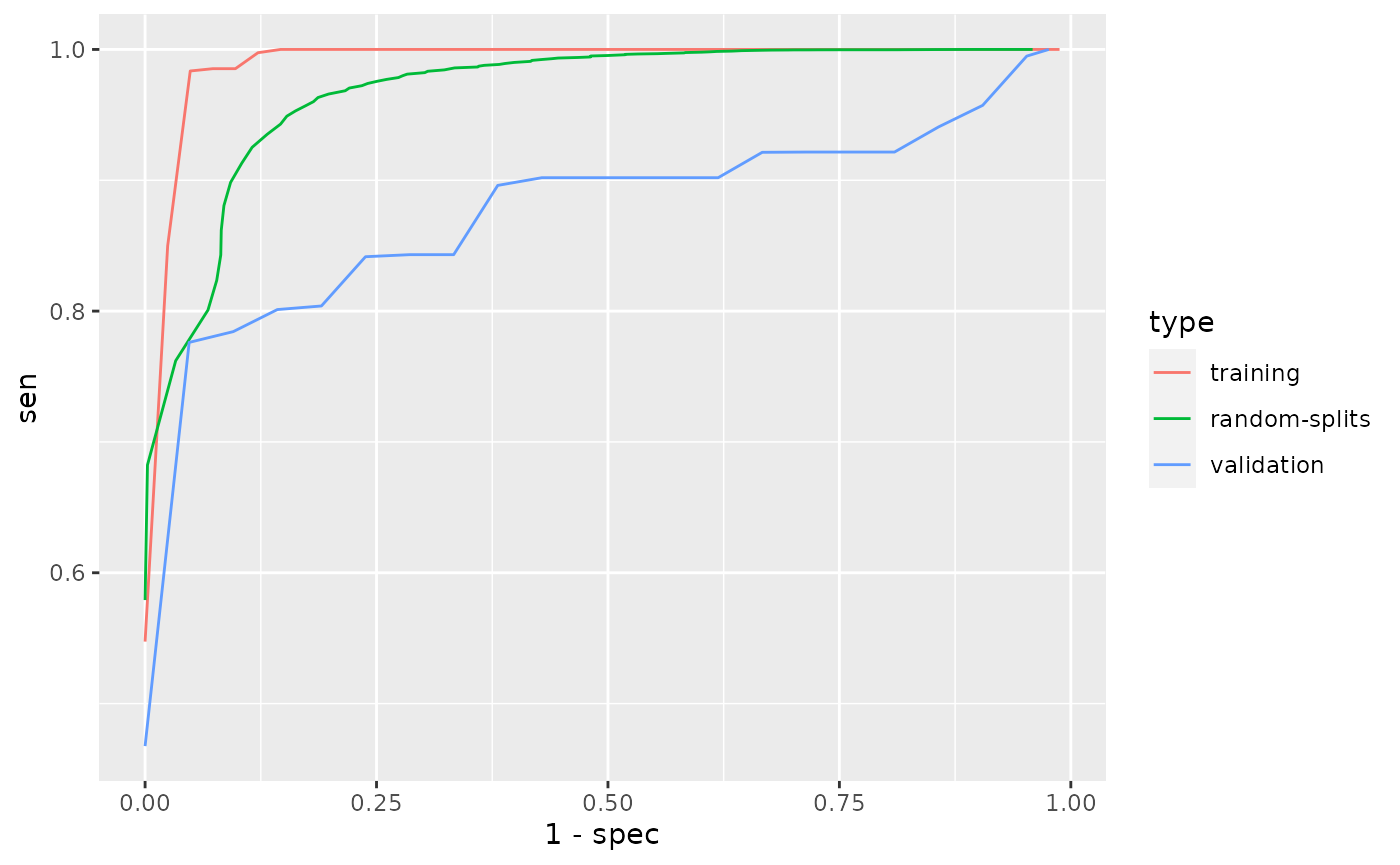
Apply the model to all the patients to obtain predicted phenotype.
phenotype <- phecap_predict_phenotype(data, model) idx <- which.min(abs(validation$valid_roc[, "FPR"] - 0.05)) cut.fpr95 <- validation$valid_roc[idx, "cutoff"] case_status <- ifelse(phenotype$prediction >= cut.fpr95, 1, 0) predict.table <- cbind(phenotype, case_status) predict.table[1:10, ]
## patient_id prediction case_status
## 1 1 1.467910e-01 0
## 2 2 9.999938e-01 1
## 3 3 1.828879e-01 0
## 4 4 9.384499e-05 0
## 5 5 1.649510e-02 0
## 6 6 9.803657e-01 1
## 7 7 8.645029e-03 0
## 8 8 2.602369e-04 0
## 9 9 3.652225e-01 0
## 10 10 7.273298e-04 0