set.seed(123)
Generate simulated data.
latent <- rgamma(8000, 0.3) latent2 <- rgamma(8000, 0.3) ehr_data <- data.frame( patient_id = 1:8000, ICD1 = rpois(8000, 7 * (rgamma(8000, 0.2) + latent) / 0.5), ICD2 = rpois(8000, 6 * (rgamma(8000, 0.8) + latent) / 1.1), ICD3 = rpois(8000, 1 * rgamma(8000, 0.5 + latent2) / 0.5), ICD4 = rpois(8000, 2 * rgamma(8000, 0.5) / 0.5), NLP1 = rpois(8000, 8 * (rgamma(8000, 0.2) + latent) / 0.6), NLP2 = rpois(8000, 2 * (rgamma(8000, 1.1) + latent) / 1.5), NLP3 = rpois(8000, 5 * (rgamma(8000, 0.1) + latent) / 0.5), NLP4 = rpois(8000, 11 * rgamma(8000, 1.9 + latent) / 1.9), NLP5 = rpois(8000, 3 * rgamma(8000, 0.5 + latent2) / 0.5), NLP6 = rpois(8000, 2 * rgamma(8000, 0.5) / 0.5), NLP7 = rpois(8000, 1 * rgamma(8000, 0.5) / 0.5), HU = rpois(8000, 30 * rgamma(8000, 0.1) / 0.1), label = NA) ii <- sample.int(8000, 400) ehr_data[ii, "label"] <- with( ehr_data[ii, ], rbinom(400, 1, plogis( -5 + 1.5 * log1p(ICD1) + log1p(NLP1) + 0.8 * log1p(NLP3) - 0.5 * log1p(HU)))) head(ehr_data)
## patient_id ICD1 ICD2 ICD3 ICD4 NLP1 NLP2 NLP3 NLP4 NLP5 NLP6 NLP7 HU label
## 1 1 2 11 3 0 0 2 2 2 1 7 0 4 NA
## 2 2 5 11 3 1 5 0 1 11 5 0 0 0 NA
## 3 3 17 10 0 2 20 2 16 10 0 0 2 0 NA
## 4 4 0 5 0 0 0 0 0 15 3 3 7 0 NA
## 5 5 0 5 1 0 0 3 7 6 31 0 3 146 NA
## 6 6 4 11 4 0 13 2 3 4 2 3 0 0 NAtail(ehr_data)
## patient_id ICD1 ICD2 ICD3 ICD4 NLP1 NLP2 NLP3 NLP4 NLP5 NLP6 NLP7 HU label
## 7995 7995 9 23 0 7 11 4 1 17 4 0 0 1 NA
## 7996 7996 21 7 9 0 6 1 1 6 4 1 2 11 NA
## 7997 7997 26 0 0 1 0 1 0 10 3 0 3 0 NA
## 7998 7998 3 0 5 1 34 1 5 21 4 4 1 46 0
## 7999 7999 0 0 0 0 4 0 0 5 0 0 5 0 NA
## 8000 8000 0 4 0 0 0 14 1 3 0 4 2 9 NADefine features and labels used for phenotyping.
data <- PhecapData(ehr_data, "HU", "label", 0.4, patient_id = "patient_id") data
## PheCAP Data
## Feature: 8000 observations of 12 variables
## Label: 132 yes, 268 no, 7600 missing
## Size of training samples: 240
## Size of validation samples: 160Specify the surrogate used for surrogate-assisted feature extraction (SAFE). The typical way is to specify a main ICD code, a main NLP CUI, as well as their combination. The default lower_cutoff is 1, and the default upper_cutoff is 10. In some cases one may want to define surrogate through lab test. Feel free to change the cutoffs based on domain knowledge.
surrogates <- list( PhecapSurrogate( variable_names = "ICD1", lower_cutoff = 1, upper_cutoff = 10), PhecapSurrogate( variable_names = "NLP1", lower_cutoff = 1, upper_cutoff = 10))
Run surrogate-assisted feature extraction (SAFE) and show result.
system.time(feature_selected <- phecap_run_feature_extraction(data, surrogates))
## user system elapsed
## 9.270 0.008 9.285
feature_selected## Feature(s) selected by surrogate-assisted feature extraction (SAFE)
## [1] "ICD1" "ICD2" "NLP1" "NLP2" "NLP3"Train phenotyping model and show the fitted model, with the AUC on the training set as well as random splits.
suppressWarnings(model <- phecap_train_phenotyping_model(data, surrogates, feature_selected)) model
## Phenotyping model:
## $lasso_bic
## (Intercept) ICD1 NLP1 HU ICD2 NLP2
## -5.3901962 1.9297295 1.2402141 -0.4471979 0.0000000 0.0000000
## NLP3
## 0.9516273
##
## AUC on training data: 0.95
## Average AUC on random splits: 0.938Validate phenotyping model using validation label, and show the AUC and ROC.
validation <- phecap_validate_phenotyping_model(data, model)
## Warning in regularize.values(x, y, ties, missing(ties)): collapsing to unique
## 'x' values
## Warning in regularize.values(x, y, ties, missing(ties)): collapsing to unique
## 'x' values
validation## AUC on validation data: 0.921
## AUC on training data: 0.95
## Average AUC on random splits: 0.938round(validation$valid_roc[validation$valid_roc[, "FPR"] <= 0.2, ], 3)
## cutoff pos.rate FPR TPR PPV NPV F1
## [1,] 0.999 0.003 0.000 0.061 1.000 0.701 0.115
## [2,] 0.969 0.100 0.009 0.308 0.939 0.759 0.464
## [3,] 0.854 0.175 0.018 0.490 0.925 0.809 0.641
## [4,] 0.680 0.194 0.027 0.584 0.907 0.837 0.710
## [5,] 0.632 0.225 0.036 0.636 0.888 0.853 0.741
## [6,] 0.596 0.253 0.050 0.700 0.864 0.874 0.773
## [7,] 0.503 0.262 0.064 0.700 0.833 0.873 0.761
## [8,] 0.461 0.269 0.073 0.700 0.814 0.872 0.753
## [9,] 0.430 0.275 0.082 0.700 0.795 0.871 0.745
## [10,] 0.408 0.281 0.091 0.700 0.778 0.870 0.737
## [11,] 0.382 0.291 0.100 0.710 0.763 0.872 0.736
## [12,] 0.341 0.300 0.109 0.720 0.750 0.875 0.735
## [13,] 0.320 0.312 0.118 0.734 0.738 0.879 0.736
## [14,] 0.297 0.331 0.127 0.772 0.734 0.894 0.752
## [15,] 0.291 0.344 0.136 0.806 0.729 0.907 0.765
## [16,] 0.290 0.359 0.150 0.820 0.713 0.912 0.763
## [17,] 0.285 0.369 0.164 0.820 0.695 0.911 0.752
## [18,] 0.255 0.375 0.173 0.824 0.684 0.912 0.748
## [19,] 0.234 0.388 0.182 0.846 0.679 0.921 0.753
## [20,] 0.215 0.400 0.191 0.868 0.674 0.931 0.759
## [21,] 0.201 0.412 0.200 0.880 0.667 0.936 0.759phecap_plot_roc_curves(validation)
## Warning: Use of `df$value_x` is discouraged. Use `value_x` instead.## Warning: Use of `df$value_y` is discouraged. Use `value_y` instead.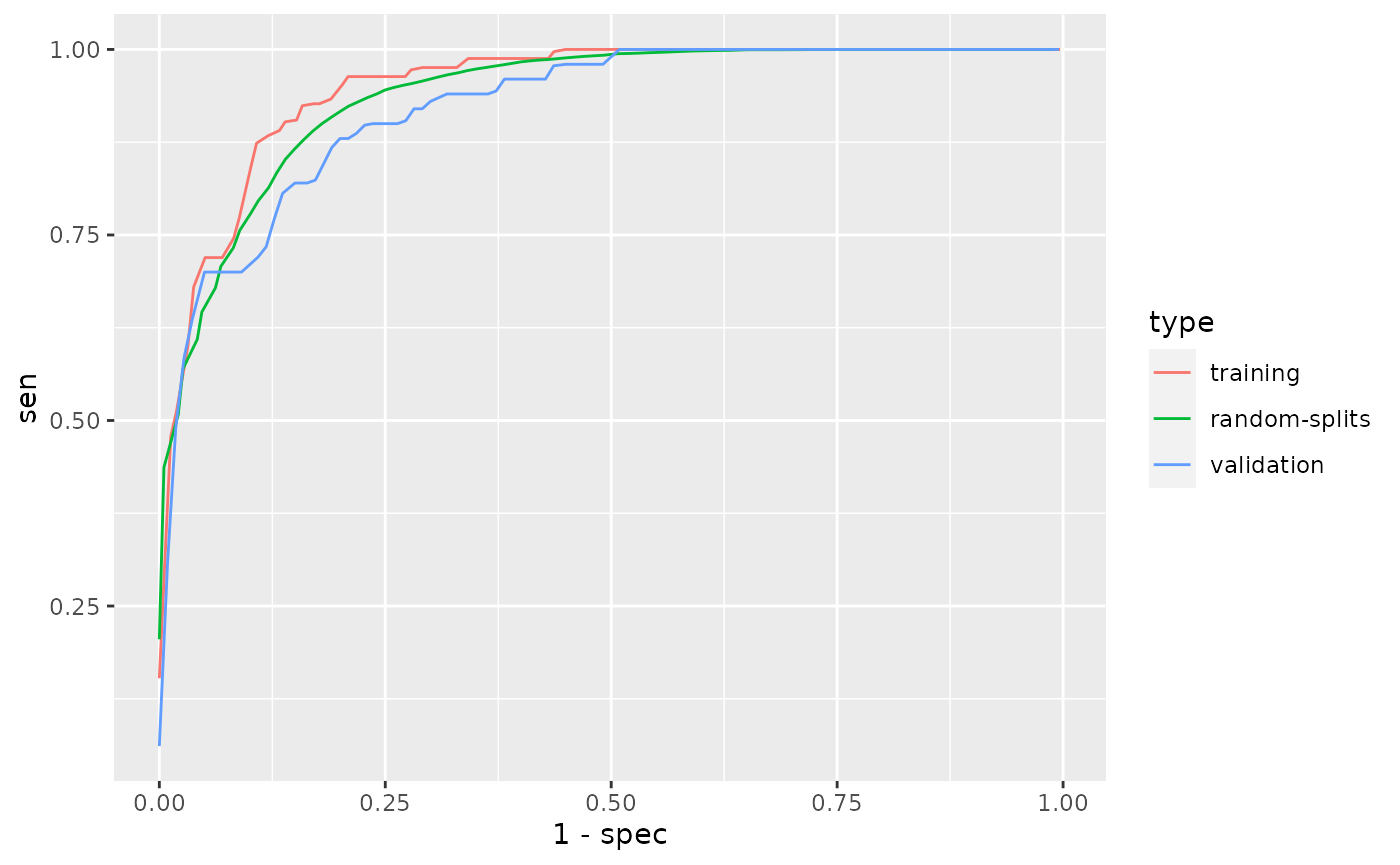
Apply the model to all the patients to obtain predicted phenotype.
phenotype <- phecap_predict_phenotype(data, model) idx <- which.min(abs(validation$valid_roc[, "FPR"] - 0.05)) cut.fpr95 <- validation$valid_roc[idx, "cutoff"] case_status <- ifelse(phenotype$prediction >= cut.fpr95, 1, 0) predict.table <- cbind(phenotype, case_status) predict.table[1:10, ]
## patient_id prediction case_status
## 1 1 0.034377662 0
## 2 2 0.433139377 0
## 3 3 0.990857331 1
## 4 4 0.004233424 0
## 5 5 0.003442046 0
## 6 6 0.702835815 1
## 7 7 0.843285354 1
## 8 8 0.001620771 0
## 9 9 0.913644435 1
## 10 10 0.044409875 0